ชุดห้องสมุด DirectFast TIANSeq (illumina)
คุณสมบัติ
■ ความสม่ำเสมอของลำดับที่ดี: ไม่มีอคติพื้นฐานของกระบวนการกระจายตัวของดีเอ็นเอและกระบวนการขยาย PCR
■ ประสิทธิภาพการแปลงห้องสมุดสูง: การสร้างห้องสมุดที่มีประสิทธิภาพสูงสามารถมั่นใจได้สำหรับตัวอย่าง DNA 1 ng
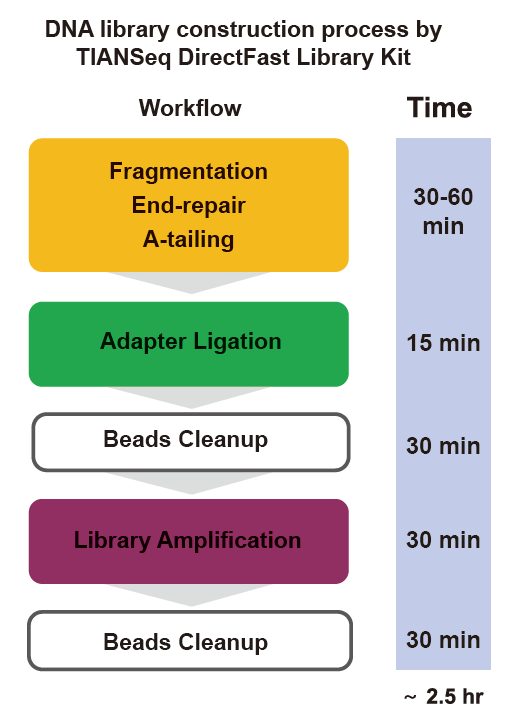
■ ดำเนินการได้รวดเร็ว: กระบวนการสร้างห้องสมุดทั้งหมดใช้เวลาเพียง 2.5 ชั่วโมงเท่านั้น
■ ประหยัดค่าใช้จ่าย: ไม่จำเป็นต้องใช้เครื่องมือและอุปกรณ์พิเศษ。
ข้อมูลจำเพาะ
พิมพ์: การเตรียมห้องสมุด DNA สำหรับแพลตฟอร์มการจัดลำดับปริมาณงานสูงของ illumina
ตัวอย่าง: จีโนม DNA หรือ DNA ชิ้นส่วนขนาดใหญ่
เป้า: ดีเอ็นเอสายคู่
เริ่มต้นอินพุตตัวอย่าง: 1 ng- 1 μg
เวลาดำเนินการ: 2.5 ชั่วโมง
แอปพลิเคชันปลายน้ำ: การจัดลำดับบนแพลตฟอร์ม illumina
ผลิตภัณฑ์ทั้งหมดสามารถปรับแต่งสำหรับ ODM/OEM สำหรับรายละเอียดกรุณาคลิกบริการที่กำหนดเอง (ODM/OEM)




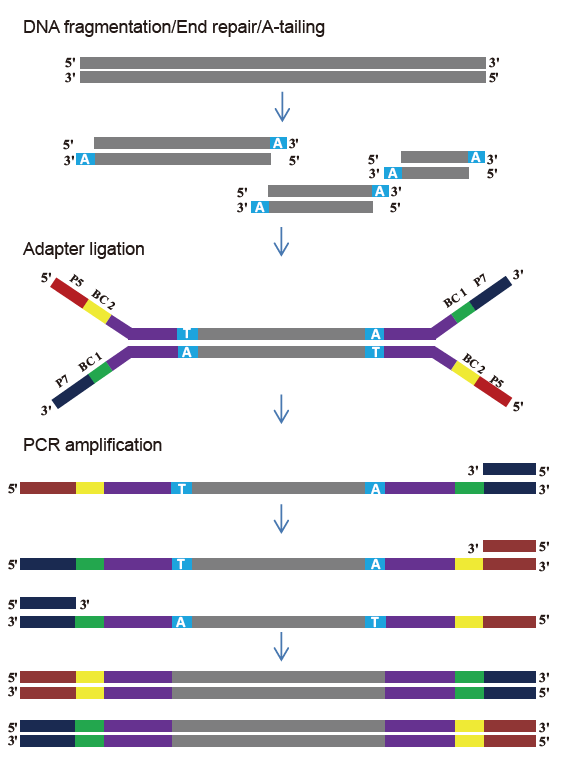
อินพุตตัวอย่างที่ยืดหยุ่นและขนาดที่แยกส่วน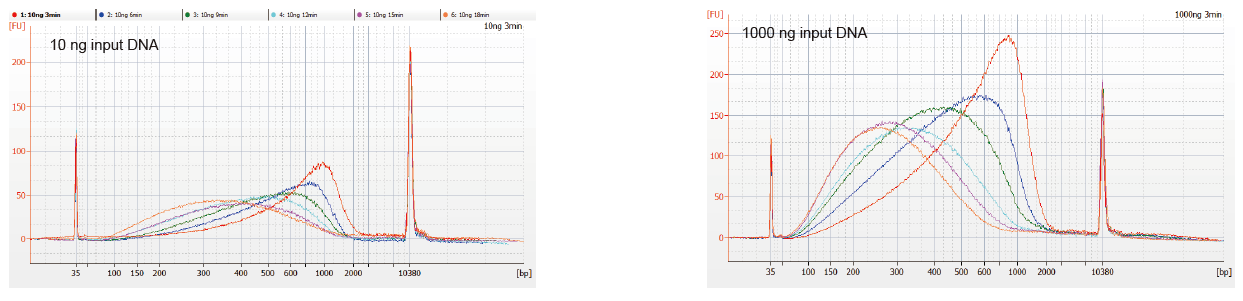 |
รูปที่ 1 โปรไฟล์การกระจายตัวของ DNA ของเวลาปฏิกิริยาที่ต่างกัน DNA 10 ng และ 1,000 ng ถูกแยกส่วนโดยใช้ TIANSeq DirectFast DNA Library Kit ผลิตภัณฑ์ปฏิกิริยาที่บำบัดด้วยเวลาปฏิกิริยาที่ต่างกันถูกทำให้บริสุทธิ์ด้วยลูกปัดแม่เหล็ก 1.8 เท่า Ampure XP และวิเคราะห์โดย Angilent 2100 |
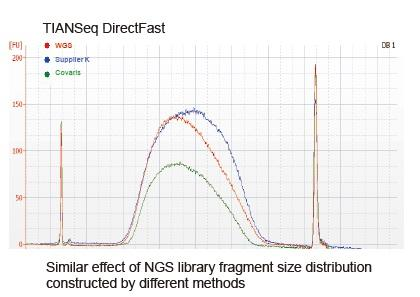
ความครอบคลุมของการจัดลำดับเหมือน Covaris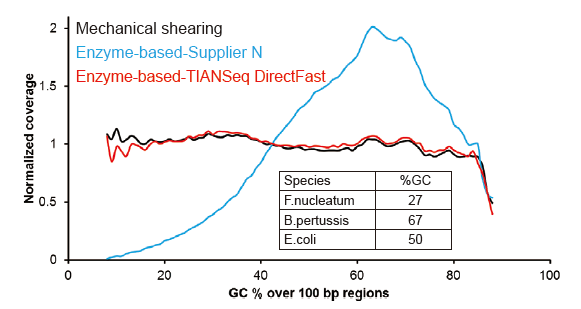 |
รูปที่ 2 การเปรียบเทียบความครอบคลุมของจีโนมของวิธีการเตรียมห้องสมุดต่างๆ จีโนม DNA ของแบคทีเรียสามตัวที่มีเนื้อหา GC ต่างกันเป็นอีโมลาร์แบบผสม และผลลัพธ์ของลำดับความครอบคลุมของจีโนมของ 100 ng ของคลัง DNA แบบผสมโดยใช้วิธีการเหล่านี้ถูกเปรียบเทียบ ผลการวิจัยพบว่า TIANSeq DirectFast Library Kit มีผลเช่นเดียวกันกับการกระจายตัวของ DNA เหมือนกับการตัดเฉือนเชิงกล และไม่มีอคติพื้นฐานสำหรับการแตกแฟรกเมนต์ |
ไม่มีอคติอย่างเป็นระบบสำหรับ DNA อินพุตต่ำเพียง 1 ng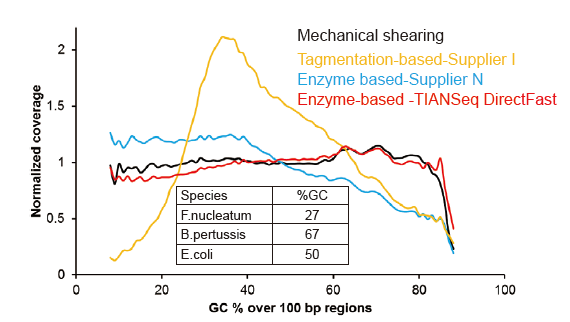 |
รูปที่ 3 การเปรียบเทียบความครอบคลุมของจีโนมของวิธีการเตรียมห้องสมุดต่างๆ จีโนม DNA ของแบคทีเรียสามตัวที่มีเนื้อหา GC ต่างกันเป็นอีโมลาร์แบบผสม และผลลัพธ์ของลำดับความครอบคลุมของจีโนมของคลัง DNA แบบผสม 1 ng โดยใช้วิธีการเหล่านี้ถูกเปรียบเทียบ ผลลัพธ์แสดงให้เห็นว่า TIANSeq DirectFast Library Kit มีผลการแตกแฟรกเมนต์ที่สม่ำเสมอด้วยการตัดเฉือนเชิงกล แม้กระทั่งสำหรับการป้อน DNA ที่ต่ำถึง 1 ng และไม่มีอคติพื้นฐาน |
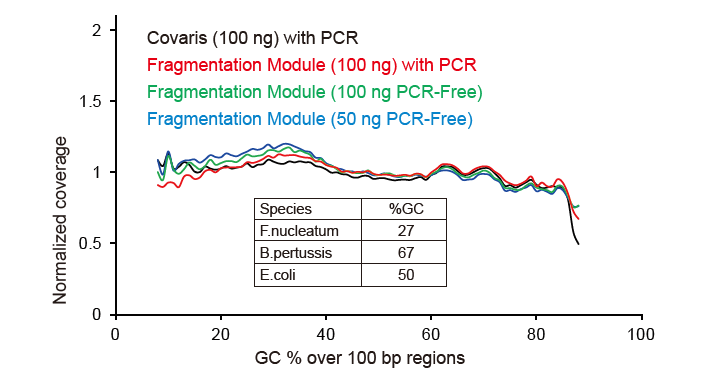
| มีความสามารถในเวิร์กโฟลว์ปลอด PCR
|
รูปที่ 4 การป้อนข้อมูลที่แตกต่างกันของ DNA จีโนมถูกใช้เพื่อสร้างห้องสมุดโดยการสร้างห้องสมุดที่ปราศจาก PCR หรือ PCR และเปรียบเทียบผลการครอบคลุมของจีโนม ผลลัพธ์แสดงให้เห็นว่าด้วยการทำงานแบบท่อเดียวและขั้นตอนการสร้างห้องสมุดที่มีประสิทธิภาพ ห้องสมุด DNA ที่สร้างด้วย TIANSeq DirectFast Library Kit ยังคงรักษาความสอดคล้องในระดับสูงด้วยการตัดเฉือนเชิงกลในการกระจายความครอบคลุมของลำดับชิ้นส่วนสำหรับเวิร์กโฟลว์ที่ปราศจาก PCR ที่เสริมด้วย PCR |
สถิติประสิทธิภาพการก่อสร้างห้องสมุดและผลผลิต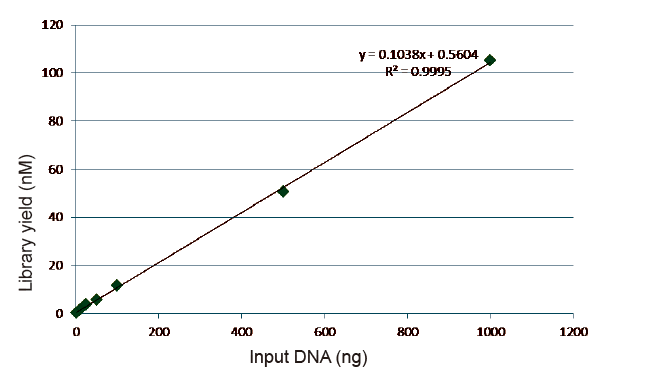 |
รูปที่ 5. ผลลัพธ์ของการวิเคราะห์เชิงปริมาณของ DNA ของห้องสมุดที่ได้รับโดย qPCR หลังจากการสร้างคลังโดยวิธีที่ปราศจาก PCR สำหรับตัวอย่างที่มีปริมาณเริ่มต้นต่างกัน (1, 10, 25, 50, 100, 500,1000 ng) การวิเคราะห์การถดถอยเชิงเส้นแสดงให้เห็นว่าผลตอบแทนของคลังมีความสัมพันธ์เชิงเส้นที่ดีในช่วงอินพุตตัวอย่างที่กว้าง สำหรับการป้อน DNA ที่ต่ำถึง 1 ng ประสิทธิภาพของการสร้างห้องสมุดจะไม่ลดลง |
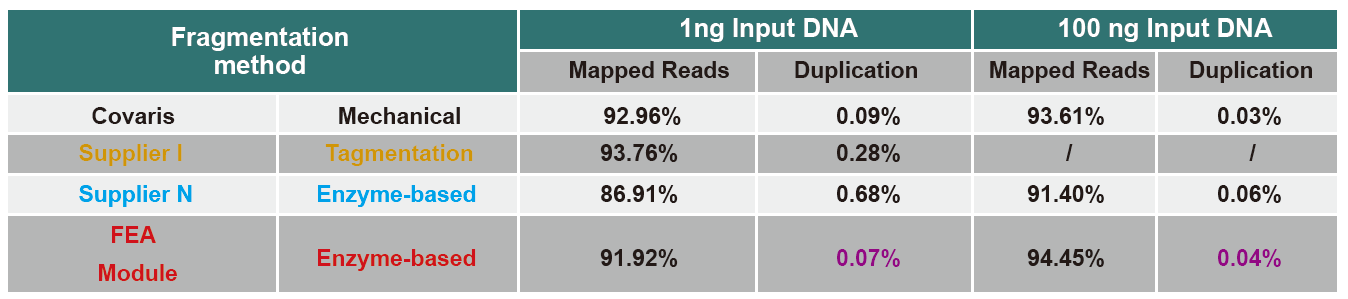
การเปรียบเทียบข้อมูลการจัดลำดับของผลิตภัณฑ์ต่างๆ
ในปัจจุบัน เทคโนโลยีการจัดลำดับปริมาณงานสูงนั้นใช้เทคโนโลยีการจัดลำดับรุ่นต่อไปเป็นหลัก เนื่องจากความยาวในการอ่านของเทคโนโลยีการจัดลำดับรุ่นถัดไปมีจำกัด เราจึงต้องแยกลำดับความยาวทั้งหมดออกเป็นไลบรารีย่อยขนาดเล็กเพื่อจัดลำดับ ตามความต้องการของการทดลองหาลำดับที่แตกต่างกัน เรามักจะเลือกการจัดลำดับแบบปลายเดียวหรือแบบปลายคู่ ปัจจุบัน ชิ้นส่วน DNA ของไลบรารีการเรียงลำดับรุ่นถัดไปโดยทั่วไปจะกระจายอยู่ในช่วง 200-800 bp
ก) DNA มีคุณภาพต่ำและมีสารยับยั้ง ใช้ตัวอย่าง DNA คุณภาพสูงเพื่อหลีกเลี่ยงการยับยั้งการทำงานของเอนไซม์
b) ปริมาณตัวอย่าง DNA ไม่เพียงพอเมื่อใช้วิธีปราศจาก PCR เพื่อสร้างคลัง DNA เมื่ออินพุตของ DNA ที่กระจัดกระจายเกิน 50 ng เวิร์กโฟลว์ที่ปราศจาก PCR สามารถเลือกดำเนินการได้ในระหว่างกระบวนการสร้างห้องสมุด ถ้าหมายเลขสำเนาของไลบรารีต่ำเกินไปที่จะจัดลำดับโดยตรง ไลบรารี DNA สามารถขยายได้โดย PCR หลังจาก ligation ของอะแด็ปเตอร์
ค) การปนเปื้อนอาร์เอ็นเอทำให้เกิดการหาปริมาณดีเอ็นเอที่ไม่ถูกต้อง การปนเปื้อนอาร์เอ็นเออาจมีอยู่ในกระบวนการทำให้บริสุทธิ์ของจีโนมดีเอ็นเอ ซึ่งอาจนำไปสู่การหาปริมาณดีเอ็นเอที่ไม่ถูกต้องและการโหลดดีเอ็นเอไม่เพียงพอระหว่างการสร้างห้องสมุด RNA สามารถลบออกได้โดยการรักษาด้วย RNase
A-1
ก) ชิ้นส่วนขนาดเล็ก (60 bp-120 bp) ปรากฏขึ้น ชิ้นส่วนขนาดเล็กมักจะเป็นชิ้นส่วนของอะแดปเตอร์หรือไดเมอร์ที่เกิดจากอะแดปเตอร์ การทำให้บริสุทธิ์ด้วยลูกปัดแม่เหล็ก Agencourt AMPure XP สามารถกำจัดชิ้นส่วนอะแดปเตอร์เหล่านี้ได้อย่างมีประสิทธิภาพและรับประกันคุณภาพการจัดลำดับ
b) แฟรกเมนต์ขนาดใหญ่ปรากฏในไลบรารีหลังการขยาย PCR ขนาดของแฟรกเมนต์ DNA ของไลบรารีจะเพิ่มขึ้น 120 bp หลังจากที่อะแด็ปเตอร์ถูกผูกมัด หากชิ้นส่วน DNA เพิ่มขึ้นมากกว่า 120 bp หลังจาก ligation ของอะแด็ปเตอร์ อาจเกิดจากการขยายชิ้นส่วนที่ผิดปกติของการขยาย PCR ที่มากเกินไป การลดจำนวนรอบ PCR สามารถป้องกันสถานการณ์ได้
c) ขนาดผิดปกติของชิ้นส่วน DNA ของห้องสมุดหลังจากเชื่อมต่ออะแดปเตอร์ ความยาวของอะแดปเตอร์ในชุดนี้คือ 60 bp เมื่อปลายทั้งสองของแฟรกเมนต์ถูกมัดเข้ากับอะแด็ปเตอร์ ความยาวจะเพิ่มขึ้นเพียง 120 bp เท่านั้น เมื่อใช้อแดปเตอร์นอกเหนือจากที่ให้มาในชุดอุปกรณ์นี้ โปรดติดต่อซัพพลายเออร์เพื่อแจ้งข้อมูลที่เกี่ยวข้อง เช่น ความยาวของอแดปเตอร์ โปรดตรวจสอบให้แน่ใจว่าขั้นตอนการทดสอบและการดำเนินการเป็นไปตามขั้นตอนที่อธิบายไว้ในคู่มือ
ง) ขนาดชิ้นส่วน DNA ผิดปกติก่อนการต่ออะแด็ปเตอร์ สาเหตุของปัญหานี้อาจเกิดจากสภาวะปฏิกิริยาที่ไม่ถูกต้องในระหว่างการแตกแฟรกเมนต์ดีเอ็นเอ ควรใช้เวลาในการตอบสนองที่แตกต่างกันสำหรับการป้อนข้อมูล DNA ที่แตกต่างกัน หากอินพุต DNA มากกว่า 10 ng เราแนะนำให้เลือกเวลาตอบสนอง 12 นาทีเป็นเวลาเริ่มต้นสำหรับการเพิ่มประสิทธิภาพ และขนาดชิ้นส่วนที่ผลิตในเวลานี้ส่วนใหญ่อยู่ในช่วง 300-500 bp ผู้ใช้สามารถเพิ่มหรือลดความยาวของชิ้นส่วนดีเอ็นเอได้เป็นเวลา 2-4 นาที ตามความต้องการของตนเองเพื่อเพิ่มประสิทธิภาพชิ้นส่วนดีเอ็นเอตามขนาดที่ต้องการ
A-2
a) เวลาในการแยกส่วนไม่ได้รับการปรับให้เหมาะสม หาก DNA ที่กระจัดกระจายมีขนาดเล็กเกินไปหรือใหญ่เกินไป โปรดดูคำแนะนำสำหรับการเลือกเวลาการแยกส่วนในคำแนะนำเพื่อกำหนดเวลาของปฏิกิริยา และใช้จุดเวลานี้เป็นตัวควบคุม ตั้งค่าเพิ่มเติม ระบบปฏิกิริยาเพื่อยืดหรือย่นเวลา 3 นาทีเพื่อให้ปรับเวลาการกระจายตัวได้แม่นยำยิ่งขึ้น
A-3
การกระจายขนาดผิดปกติของ DNA หลังการรักษาการกระจายตัว
ก) วิธีการละลายของรีเอเจนต์ที่แตกตัวไม่ถูกต้อง หรือรีเอเจนต์ไม่ได้ผสมอย่างสมบูรณ์หลังจากการละลาย ละลายรีเอเจนต์ 5 เท่าของ Fragmentation Enzyme Mix บนน้ำแข็ง เมื่อละลายแล้ว ให้ผสมรีเอเจนต์เท่าๆ กันโดยสะบัดเบาๆ ที่ก้นหลอด อย่าวนรีเอเจนต์!
b) ตัวอย่างอินพุต DNA มี EDTA หรือสารมลพิษอื่นๆ การทำให้เกลือไอออนและสารคีเลตหมดลงในขั้นตอนการทำให้บริสุทธิ์ของ DNA นั้นมีความสำคัญอย่างยิ่งต่อความสำเร็จของการทดลอง ถ้า DNA ละลายใน 1×TE ให้ใช้วิธีการที่ให้ไว้ในคำแนะนำเพื่อทำการแยกส่วน หากความเข้มข้นของ EDTA ในสารละลายไม่แน่นอน ขอแนะนำให้ทำให้ DNA บริสุทธิ์และละลายในน้ำที่ปราศจากไอออนสำหรับปฏิกิริยาต่อไป
c) การหาปริมาณ DNA เริ่มต้นที่ไม่ถูกต้อง ขนาดของ DNA ที่แยกส่วนมีความสัมพันธ์อย่างใกล้ชิดกับปริมาณของ DNA ที่ป้อนเข้าไป ก่อนการรักษาการกระจายตัว การหาปริมาณ DNA ที่แม่นยำโดยใช้ Qubit, Picogreen และวิธีการอื่นๆ เป็นสิ่งสำคัญในการกำหนดปริมาณ DNA ที่แน่นอนในระบบปฏิกิริยา
ง) การเตรียมระบบปฏิกิริยาไม่เป็นไปตามคำแนะนำ การเตรียมระบบปฏิกิริยาแบบแยกส่วนต้องดำเนินการบนน้ำแข็งอย่างเคร่งครัดตามคำแนะนำ เพื่อให้ได้ผลลัพธ์ที่ดีที่สุด ควรวางส่วนประกอบปฏิกิริยาทั้งหมดบนน้ำแข็ง และเตรียมระบบปฏิกิริยาหลังจากเย็นตัวลงอย่างสมบูรณ์ หลังจากเตรียมเสร็จ โปรดสะบัดหรือปิเปตให้คลุกเคล้าให้เข้ากัน อย่ากระแสน้ำวน!
1. วิธีการผสมที่ไม่เหมาะสม (กระแสน้ำวน การแกว่งอย่างรุนแรง ฯลฯ) จะทำให้การกระจายส่วนย่อยของห้องสมุดผิดปกติ (ดังแสดงในรูปต่อไปนี้) ส่งผลต่อคุณภาพของห้องสมุด ดังนั้น เมื่อเตรียมสารละลายปฏิกิริยา Fragmentation Mix โปรดปิเปตขึ้นและลงเบา ๆ เพื่อผสม หรือใช้ปลายนิ้วปัดและผสมให้เข้ากัน ระวังอย่าผสมกับกระแสน้ำวน
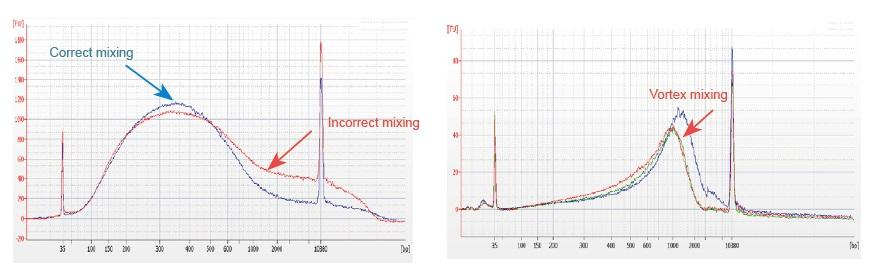
2. ต้องใช้ DNA ที่มีความบริสุทธิ์สูงในการสร้างห้องสมุด
■ ความสมบูรณ์ของ DNA ที่ดี: แถบอิเล็กโตรโฟรีซิสมีขนาดมากกว่า 30 kb โดยไม่มีหาง
■ OD260/230: >1.5
■ OD260/280: 1.7-1.9
3. ปริมาณ DNA ที่ป้อนต้องแม่นยำ แนะนำให้ใช้วิธี Qubit และ PicoGreen ในการหาปริมาณ DNA มากกว่า Nanodrop
4. ต้องกำหนดเนื้อหาของ EDTA ในสารละลาย DNA EDTA มีอิทธิพลอย่างมากต่อปฏิกิริยาการแตกตัว หากเนื้อหาของ EDTA สูง จะต้องทำการทำให้ DNA บริสุทธิ์ก่อนการทดสอบครั้งต่อไป
5. สารละลายปฏิกิริยาการแตกตัวต้องเตรียมบนน้ำแข็ง กระบวนการแตกตัวนั้นไวต่ออุณหภูมิและเวลาของปฏิกิริยา เพื่อความถูกต้องของเวลาปฏิกิริยา โปรดเตรียมระบบปฏิกิริยาบนน้ำแข็ง
6. เวลาปฏิกิริยาการแตกแฟรกเมนต์ต้องแม่นยำ เวลาตอบสนองของขั้นตอนการแตกแฟรกเมนต์จะส่งผลโดยตรงต่อขนาดของผลิตภัณฑ์จากแฟรกเมนต์ ซึ่งส่งผลต่อการกระจายขนาดของชิ้นส่วนดีเอ็นเอในห้องสมุด
1. ตัวอย่างประเภทใดที่เหมาะกับชุดนี้?
ชนิดตัวอย่างที่ใช้ได้ของชุดอุปกรณ์นี้สามารถเป็น RNA ทั้งหมดหรือ mRNA ที่บริสุทธิ์และมีความสมบูรณ์ของ RNA ที่ดี หากใช้ RNA ทั้งหมดเพื่อสร้างไลบรารี ขอแนะนำให้ใช้ชุดการแยก rRNA (Cat#4992363/4992364/4992391) เพื่อลบ rRNA ก่อน
2. ตัวอย่าง FFPE สามารถใช้สร้างห้องสมุดด้วยชุดนี้ได้หรือไม่?
mRNA ในตัวอย่าง FFPE จะถูกลดระดับลงในระดับหนึ่ง โดยมีความสมบูรณ์ค่อนข้างต่ำ เมื่อใช้ชุดนี้สำหรับการสร้างห้องสมุด ขอแนะนำให้ปรับเวลาการแตกแฟรกเมนต์ให้เหมาะสม
3. ใช้ขั้นตอนการเลือกขนาดที่ให้มาในคู่มือผลิตภัณฑ์ อะไรอาจทำให้ส่วนที่แทรกดูเบี่ยงเบนเล็กน้อย
การเลือกขนาดจะต้องดำเนินการอย่างเคร่งครัดตามขั้นตอนการเลือกขนาดในคู่มือผลิตภัณฑ์นี้ หากมีการเบี่ยงเบน สาเหตุอาจเป็นเพราะเม็ดแม่เหล็กไม่สมดุลกับอุณหภูมิห้องหรือผสมกันไม่เต็มที่ ปิเปตไม่ถูกต้อง หรือของเหลวยังคงอยู่ในปลาย ขอแนะนำให้ใช้เคล็ดลับที่มีการดูดซับต่ำสำหรับการทดลอง
4. การเลือกอะแดปเตอร์ในการสร้างห้องสมุด
ชุดสร้างไลบรารีไม่มีรีเอเจนต์อะแดปเตอร์ และขอแนะนำให้ใช้ชุดนี้ร่วมกับอะแดปเตอร์ TIANSeq Single-Index (Illumina) (4992641/4992642/4992378)
5. QC ของห้องสมุด
การตรวจจับเชิงปริมาณของไลบรารี: Qubit และ qPCR ใช้เพื่อกำหนดความเข้มข้นของมวลและความเข้มข้นของโมลาร์ของไลบรารีตามลำดับ การดำเนินการเป็นไปตามคู่มือผลิตภัณฑ์อย่างเคร่งครัด ความเข้มข้นของห้องสมุดโดยทั่วไปจะเป็นไปตามข้อกำหนดของการจัดลำดับ NGS การตรวจจับช่วงการแจกจ่ายห้องสมุด: การใช้ Agilent 2100 Bioanalyzer เพื่อตรวจหาช่วงการแจกจ่ายห้องสมุด
6. การเลือกหมายเลขรอบการขยาย
ตามคำแนะนำ จำนวนรอบ PCR คือ 6-12 และควรเลือกจำนวนรอบ PCR ที่ต้องการตามอินพุตตัวอย่าง ในไลบรารีที่ให้ผลตอบแทนสูง การขยายสัญญาณเกินมักจะเกิดขึ้นในระดับที่แตกต่างกัน ซึ่งแสดงให้เห็นโดยจุดสูงสุดที่ใหญ่กว่าเล็กน้อยหลังจากจุดสูงสุดของช่วงเป้าหมายในการตรวจจับ Agilent 2100 Bioanalyzer หรือความเข้มข้นที่ตรวจพบของ Qubit ต่ำกว่าของ qPCR การขยายสัญญาณแบบอ่อนเกินไปเป็นปรากฏการณ์ปกติ ซึ่งไม่ส่งผลต่อการจัดลำดับไลบรารีและการวิเคราะห์ข้อมูลที่ตามมา
7. Spikes ปรากฏในโปรไฟล์การตรวจจับของ Agilent 2100 Bioanalyzer
การปรากฏตัวของหนามแหลมในการตรวจจับ Agilent 2100 Bioanalyzer นั้นเกิดจากการแยกส่วนอย่างไม่สม่ำเสมอของตัวอย่าง ซึ่งจะมีชิ้นส่วนมากขึ้นในบางขนาด และสิ่งนี้จะชัดเจนมากขึ้นหลังจากการเสริม PCR ในกรณีนี้ ขอแนะนำว่าอย่าทำการเลือกขนาด กล่าวคือ ตั้งค่าสภาวะการกระจายตัวเป็น 94°C เป็นเวลา 15 นาทีในการฟักไข่ โดยที่การกระจายของแฟรกเมนต์มีขนาดเล็กและเข้มข้น และสามารถปรับปรุงความเป็นเนื้อเดียวกันได้
หมวดหมู่สินค้า
ทำไมถึงเลือกพวกเรา
นับตั้งแต่ก่อตั้ง โรงงานของเราได้พัฒนาผลิตภัณฑ์ระดับโลกเป็นครั้งแรกโดยยึดมั่นในหลักการ
ที่มีคุณภาพก่อน ผลิตภัณฑ์ของเราได้รับชื่อเสียงที่ดีเยี่ยมในอุตสาหกรรมและมีคุณค่าความไว้วางใจจากลูกค้าทั้งเก่าและใหม่..
- โทร: +86 010-59822688
- อาคาร 5 เลขที่ 86 ถนน Shuangying West เขต Changping ปักกิ่ง
- people@tiangen.com



